文章目录
- Deep learning at base-resolution reveals cis-regulatory motif syntax
- problem
- BPNet: predicting base-resolution profiles from DNA sequence
- Interpreting the predictions of BPNet
- 1 DeepLIFT
- 2 TF-MoDISCO
- 3 motif syntax derived TF cooperativity
- Experimental validation of BPNet predictions
- summary
来自Manolis Kellis教授(MIT计算生物学主任)的课
油管链接:Regulatory Genomics - Deep Learning in Life Sciences - Lecture 07 (Spring 2021)
本节课分为三个部分,本篇笔记是第二部分。
本节主要是请了一个阿三哥来介绍来一下他们的一些将神经网络应用在调控基因组学上的工作,模型的原理、意义和拓展。模型叫BPNet、DeepLIFT以及TF-MoDISCO。并且在实验上验证了他。整体思路递进的还不错,但是讲的有点快和凌乱,我感觉不如下一部分英伟达的讲座
Deep learning at base-resolution reveals cis-regulatory motif syntax
26:07
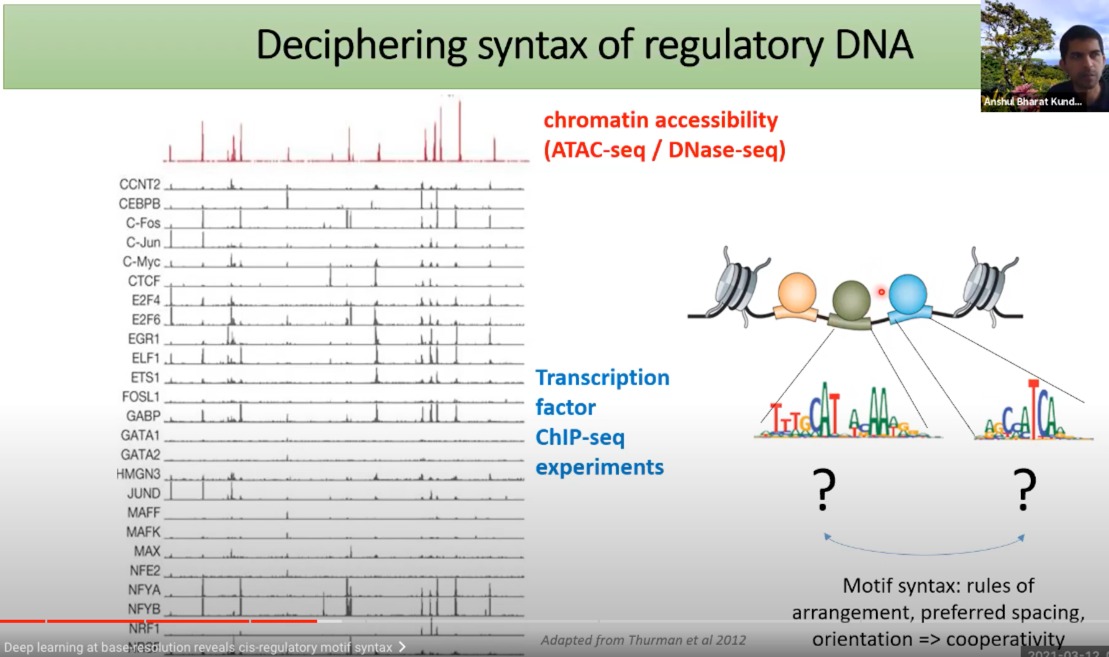
Motif syntax 是描述转录因子或其他DNA结合蛋白质如何识别和结合它们的目标DNA motif的“规则”。
- Arrangement(排列):motif中的特定核苷酸序列的顺序。
- Preferred Spacing(优选间距):当多个转录因子结合到相近的位置时,它们之间的距离可以影响它们的结合和功能。这里的间距指的是两个motif或两个结合位点之间的核苷酸数。
- Orientation(方向性):转录因子可以以不同的方向结合到DNA上。某些转录因子可能优先在某个特定的方向上结合,而其他的可能不在乎。
- Cooperativity(协同性):这是指当一个转录因子结合到其目标motif时,它可能会影响其他转录因子与附近的DNA结合的能力。例如,一个转录因子的结合可能会使另一个转录因子更容易(或更困难)结合到附近的位置。
- 调控性DNA的预测模型
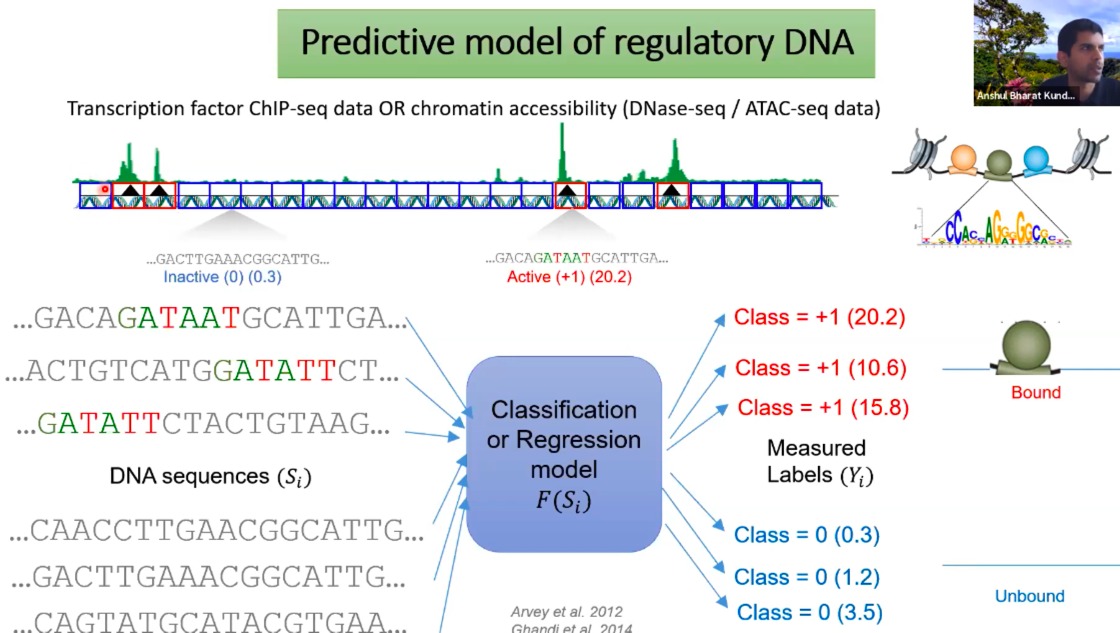
-
Transcription factor ChIP-seq data OR chromatin accessibility (DNase-seq / ATAC-seq data): 这一部分展示了两种主要的高通量测序数据——转录因子的ChIP-seq数据和染色质的可及性数据(如DNase-seq和ATAC-seq)。
- 绿色的峰值表示转录因子在基因组上的结合位置或染色质在特定位置的可及性。
-
中部的DNA序列:
- 基因组的特定片段。每一个片段都有一个对应的标签,这个标签可能代表该片段是否活跃(例如,是否有转录因子结合)及其活跃程度。
- 例如,“Inactive (0) (0.3)”表示这个片段是不活跃的,而“Active (+1) (20.2)”则表示它是活跃的,并且活跃度为20.2。
-
Classification or Regression model F(Si):
- 核心部分。这个模型接受DNA序列片段作为输入,并预测它们的活跃状态或其他某种测量值。这可以是一个分类问题(例如,预测一个给定的DNA片段是否活跃)或一个回归问题(例如,预测其活跃程度或转录因子结合的强度)。
-
Measured Labels (Yi):
- 这些是真实的、通过实验得到的数据标签。
通过DNA序列信息来预测基因表达的调控模式,特别是转录因子如何与基因组互动。这种模型有助于我们理解基因如何被调控,以及哪些因素可能影响基因的活跃状态。
这些用来训练的片段好像是固定长度的?
我有一些疑惑:
这里训练的数据的x就是dna序列信息,预测出来的就是1或者0,好像没用到motif的信息啊
从图中所示的模型结构来看,确实直接使用了DNA序列作为输入,并预测出一个分类结果(例如1或0,表示转录因子是否结合)。但是,即使模型直接使用DNA序列作为输入,Motif信息仍然在以下方式中隐式地起到作用:
特征提取: 即使模型的直接输入是DNA序列,它内部的机制可能会学习到与Motif相似的特征或模式。例如,如果使用深度学习模型,如卷积神经网络(CNN),网络的卷积层可能会识别出与Motif相关的序列模式。
标签来源: 虽然模型预测的输出是1或0,但这些标签可能基于实验数据,如ChIP-seq数据。ChIP-seq实验可以确定转录因子在基因组上的确切结合位置,这些位置往往与特定的Motif相关。
后处理与解释: 一旦模型进行了训练并进行预测,研究者可能会进一步分析模型识别的特征或模式,以理解哪些DNA Motif与预测的结合事件相关。
problem
这种预测方式实际上丢失了很多信息
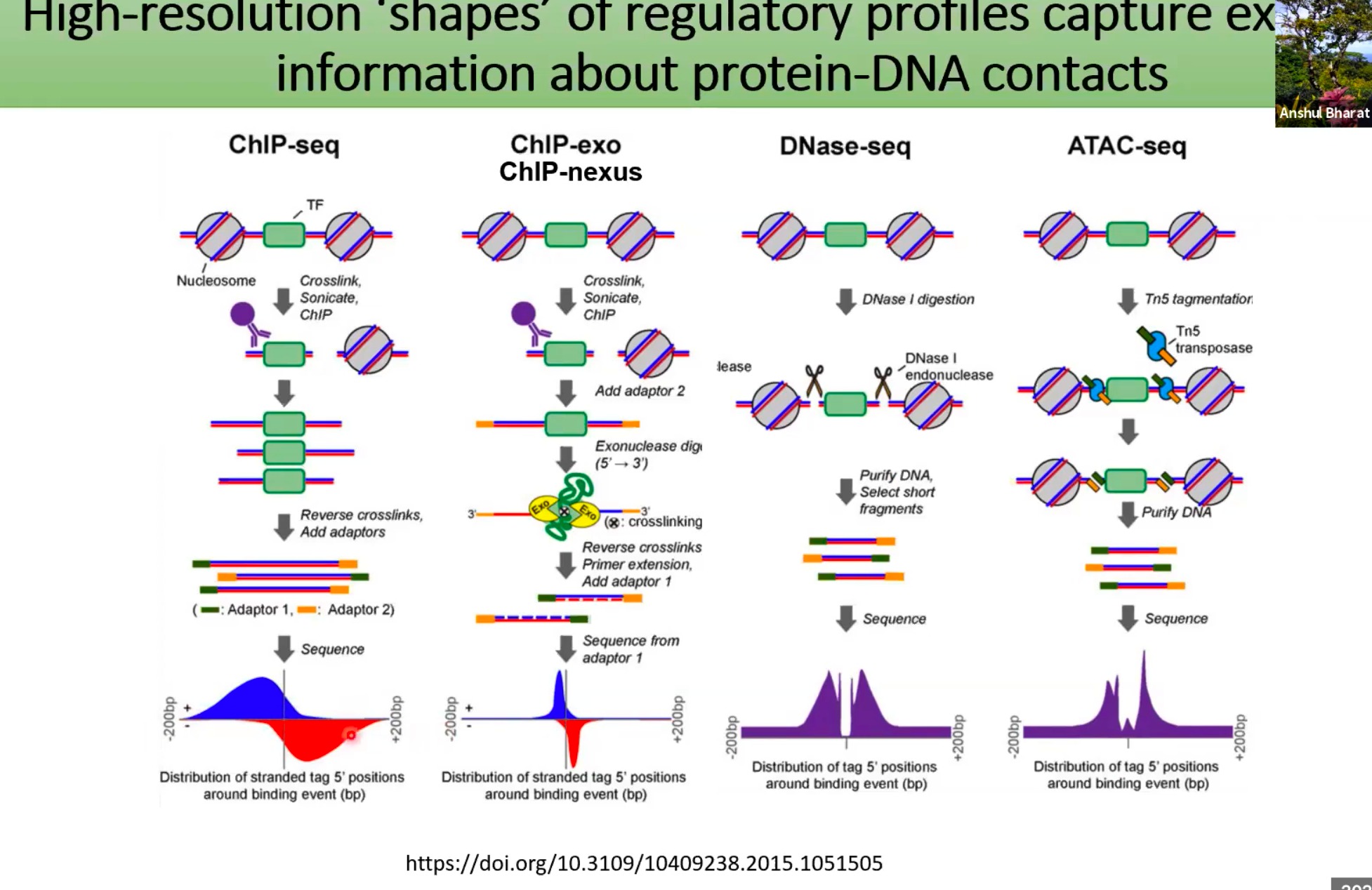
使用高分辨率技术来捕捉蛋白质与DNA接触的详细信息
简单介绍一下四种技术:
- ChIP-seq:
- 首先,目标蛋白质(例如转录因子,简称TF)与DNA结合。接下来,通过交联、超声波处理和ChIP(染色体免疫沉淀)将这些结合的区域隔离出来。
- 结合的DNA被逆交联并加入适配子,然后进行测序。测序结果显示为在结合事件周围的分布。
- ChIP-exo/ChIP-nexus:
- 这与ChIP-seq相似,但在测序前有额外的步骤。特定的适配子被加入,然后进行外切酶消化。
- 结果提供了更高的空间分辨率,能够更精确地确定蛋白质与DNA的接触点。
- DNase-seq
- ATAC-seq:
- 这里就产生问题了,当我们使用如ChIP-seq、ATAC-seq等技术时,我们能够获得大量关于DNA和蛋白质之间相互作用的信息。
- 但前面的模型将这些数据简单的转化为单一的标量值(例如某个特定区域的总读数),可能会损失大量的细节信息。这些细节可能包括DNA上特定的结构特征、绑定模式(motif)的语法等,这些都是非常有价值的信息。
类比一下:
假设DNA像一本书的长篇章节,而蛋白质则像是标记章节中某些关键词或句子的高光笔。通过技术如ChIP-seq和ATAC-seq,我们可以确定蛋白质“标记”了哪些部分的DNA。这就好比看到哪些句子或关键词被高光笔标记了。
现在,**如果我们只关心每页被标记的次数,而不关心具体标记了哪些句子或关键词,我们就会丢失很多信息。**在DNA的情境中,这意味着我们只关心某个区域有多少蛋白质绑定,而不关心具体是哪些位置上的DNA与蛋白质绑定。
但DNA上的每一个位置都很重要,因为它们可以告诉我们哪些部分的DNA更可能控制某些功能。这就好像知道哪些句子或关键词被高光笔标记可以告诉我们这本书的主题和内容。
另外,DNA上的motif会以特定的方式与蛋白质互动。这些序列的语法和结构就好比语言中的语法规则。如果我们忽略了这些信息,我们可能会错过理解DNA如何工作的关键部分。
BPNet: predicting base-resolution profiles from DNA sequence
从DNA序列预测基对分辨率的数据。
该模型的目标是能够准确地预测蛋白质与DNA之间的具体交互位置,而不仅仅是整体的交互强度。
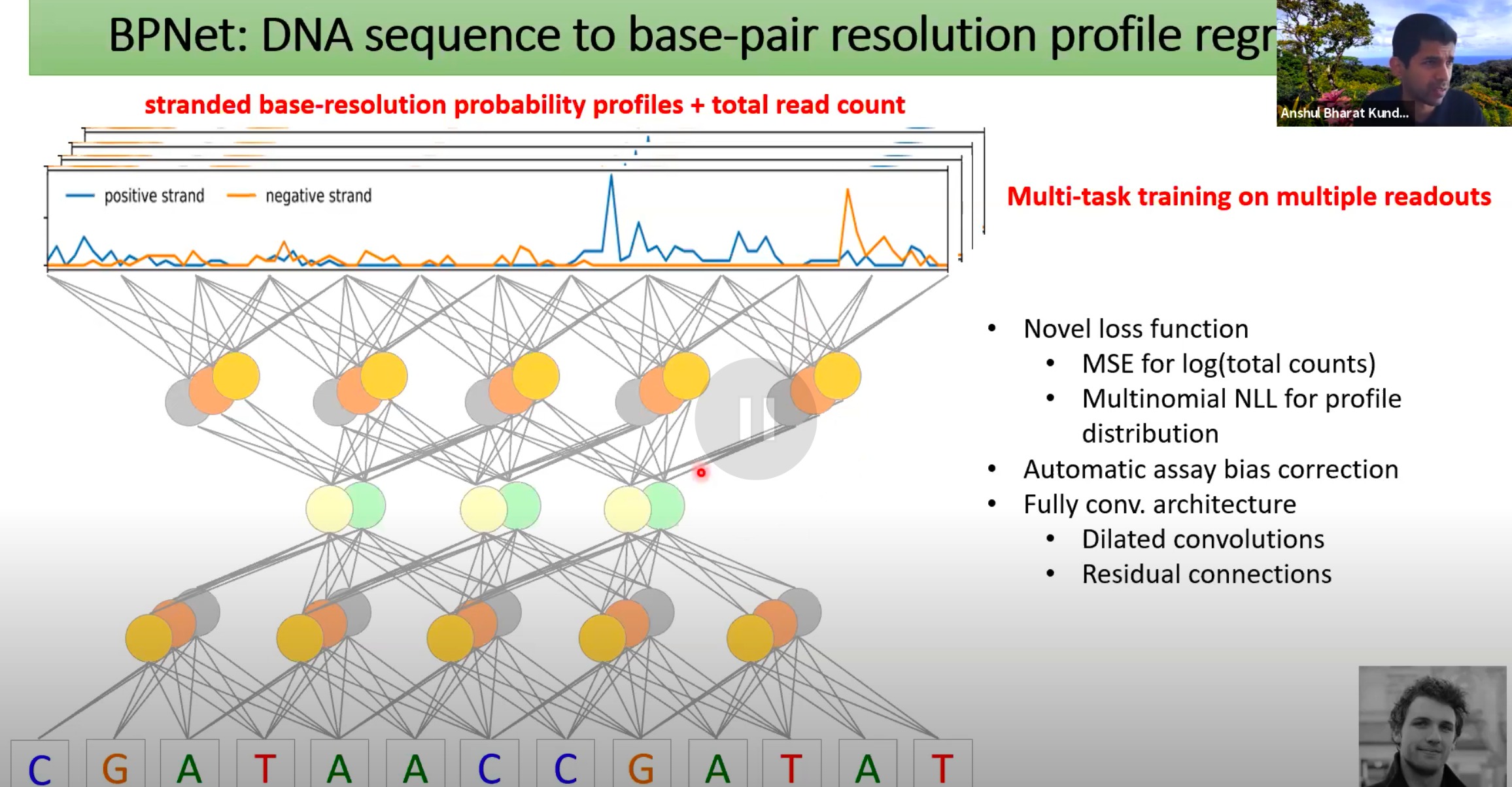
profile就是一组相关序列的共同特征,我认为这里就是指转录因子(蛋白质)在一条DNA上位置的结合强度
-
模型能够为DNA的正链和负链预测与蛋白质交互的概率
- base-pair,每一个碱基对位置与蛋白质交互的概率
- 说明训练的label数据有更多的细节,更高的分辨率,而不是简单的凑合成一个标量
-
Multi-task
- 模型能够同时预测多种输出
- 对于chip-seq实验来说,相同的序列可能对应四五种不同蛋白质有不同的概率轮廓结果
-
loss function
-
MSE for log(total counts):使用均方误差预测整体读数的对数值。
-
Multinomial NLL for profile distribution:使用多项式负对数似然预测与蛋白质交互的具体位置。
-
-
自动纠正偏差
- 某些实验方法可能会引入偏差,导致某些区域的交互数据被放大或减小
-
Dilated convolutions and Residual connections:这些是深度学习模型中的高级技术,可以帮助模型捕获数据中的长距离依赖性并提高训练速度。
模型比较简单,阿三哥认为模型最创新的地方不是网络结构,而是损失函数的设计。不要二元预测就逻辑回归,不要数值预测就均方误差。要学会根据数据中的噪声的性质来设计损失函数。
他认为Multinomial NLL这种分布更加适合几千个碱基对的序列中每个读数的精确分布情况
- (这里我也不是太明白,我需要去理解一下整体的生物实验与其结果是啥),期待未来补充
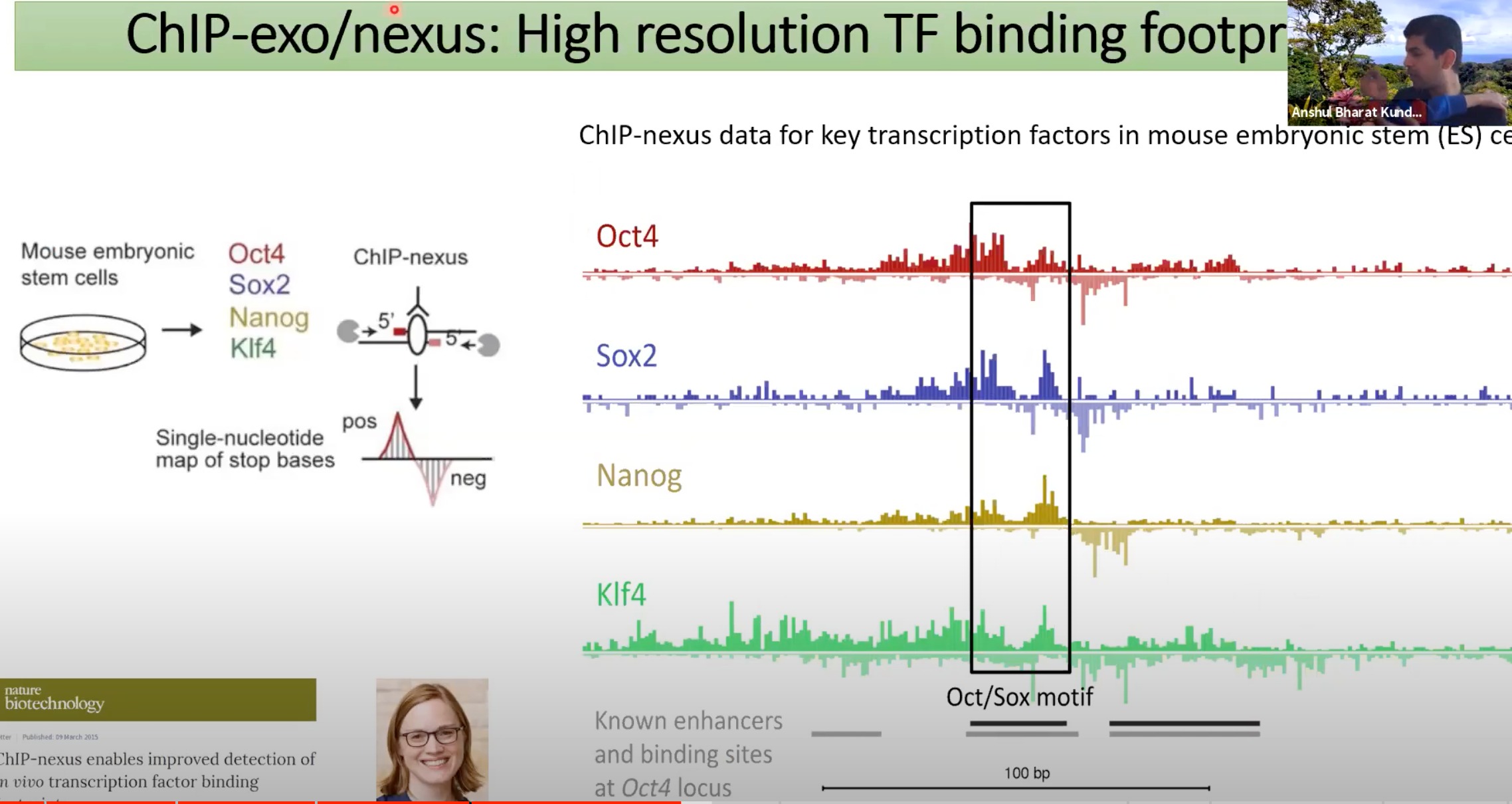
-
ChIP-exo/nexus是一种技术,可以用来检测转录因子(TF)结合到DNA的精确位置
-
四个小鼠胚胎干细胞中关键的转录因子:Oct4, Sox2, Nanog, 和 Klf4
-
预测结果
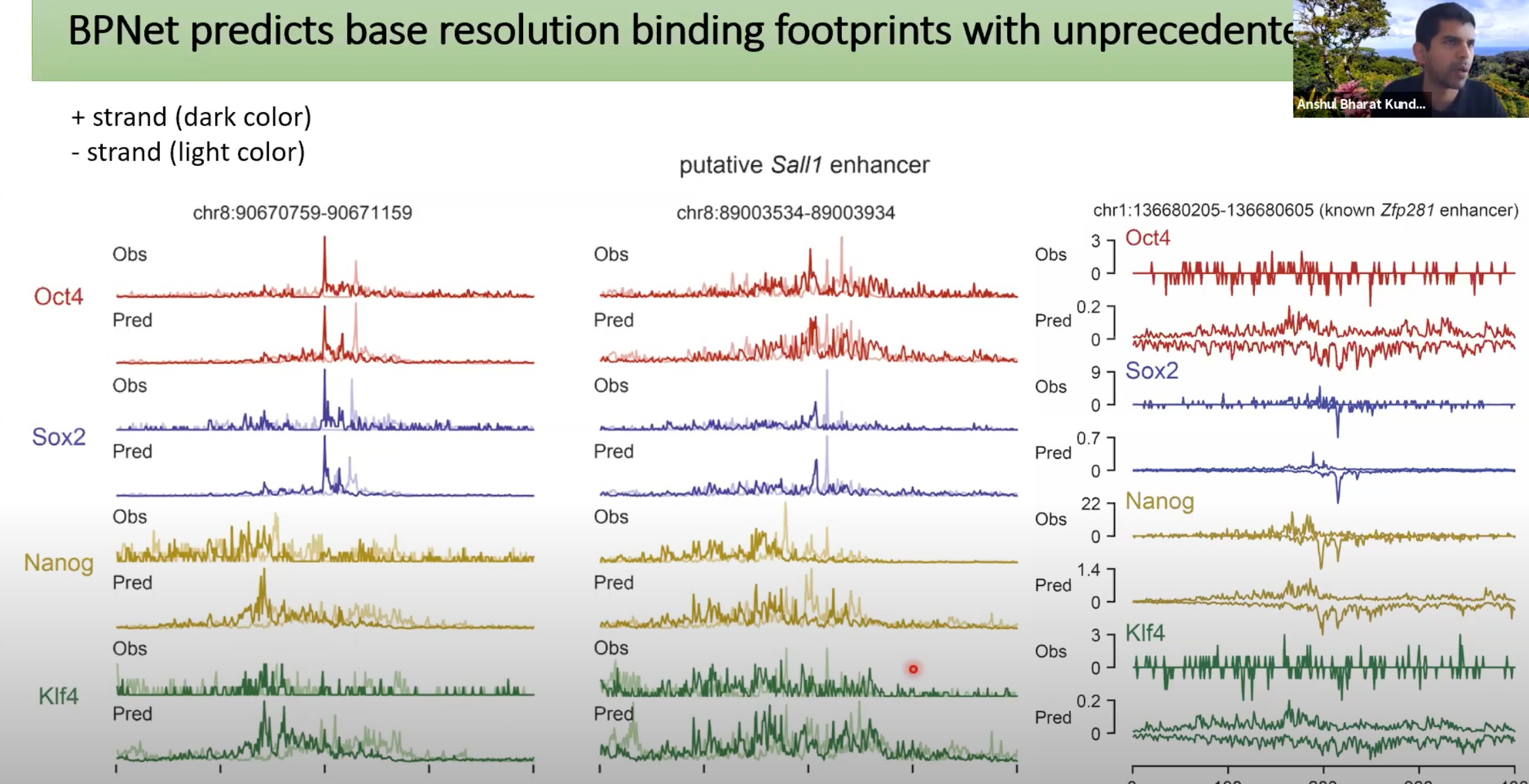
可以看出预测的很好,最右边是因为这个数据本身就有些缺失值
图中三个区域是不同的三个DNA区域
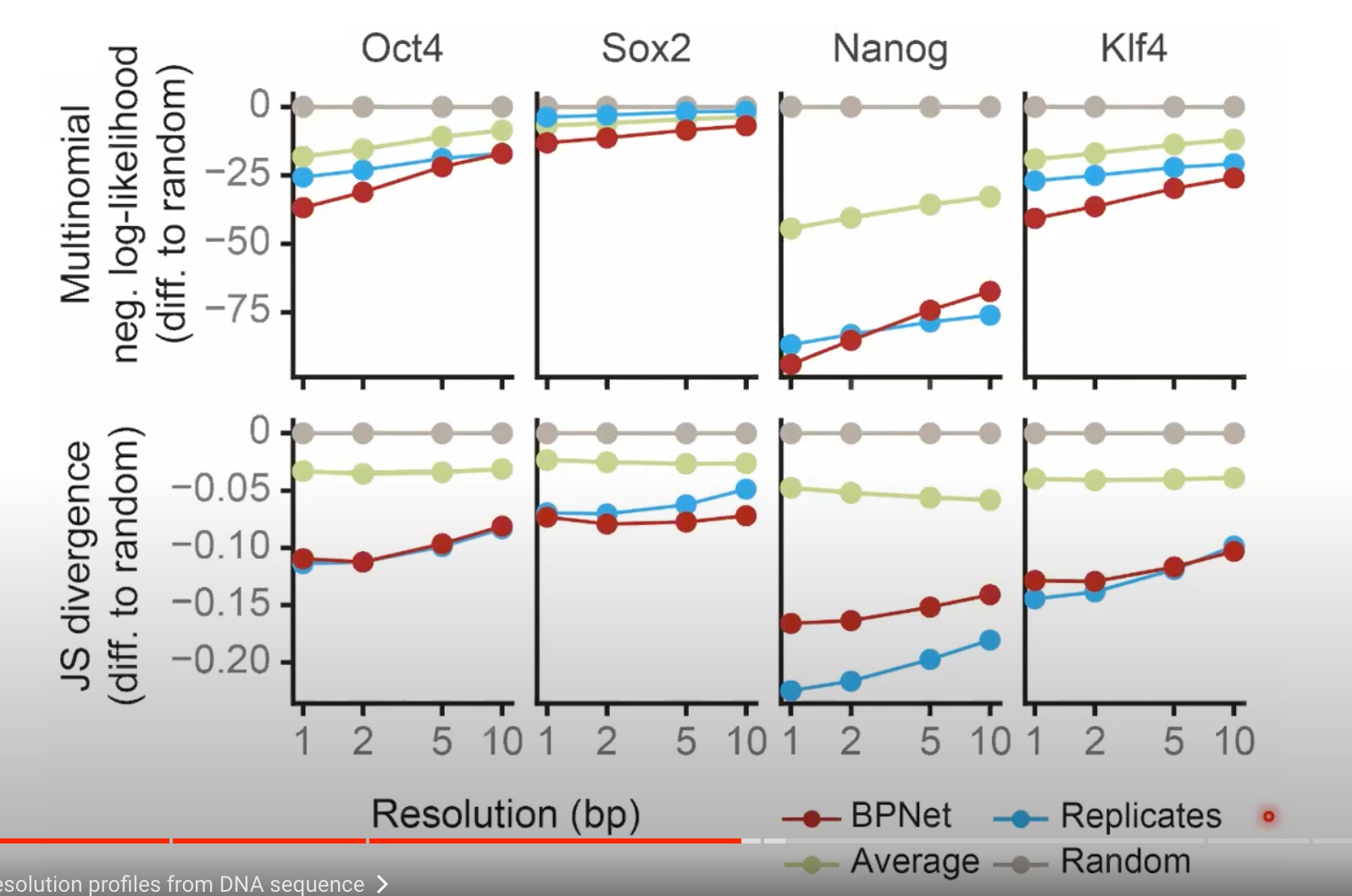
-
第一行是指越低预测越好
-
第二行是Jensen-Shannon散度,衡量两个概率分布之间相似性的一个方法。值也是越低越好
-
这里不同的resolution,分辨率,应该是对结果计算时进行了一定的平滑处理,比如5bp的分辨率就是在评估预测性能的时候,将每5bp的预测值取均值后再进行比较
- 可以减少噪声并更清楚地展示趋势
-
红线和蓝线越接近,模型的预测能力越接近实验数据,效果越好
Interpreting the predictions of BPNet
Deciphering predictive motifs and motif instances
终于来到重点了,我还寻思这预测什么用,实验数据都能做出来了。这确实没有什么用,重要的是根据碱基序列中的motifs如何能影响不同位置上蛋白质结合强度的预测,其背后到底发生了什么。
1 DeepLIFT
Deep Learning Important FeaTures,目标是确定输入特征对于模型预测的贡献
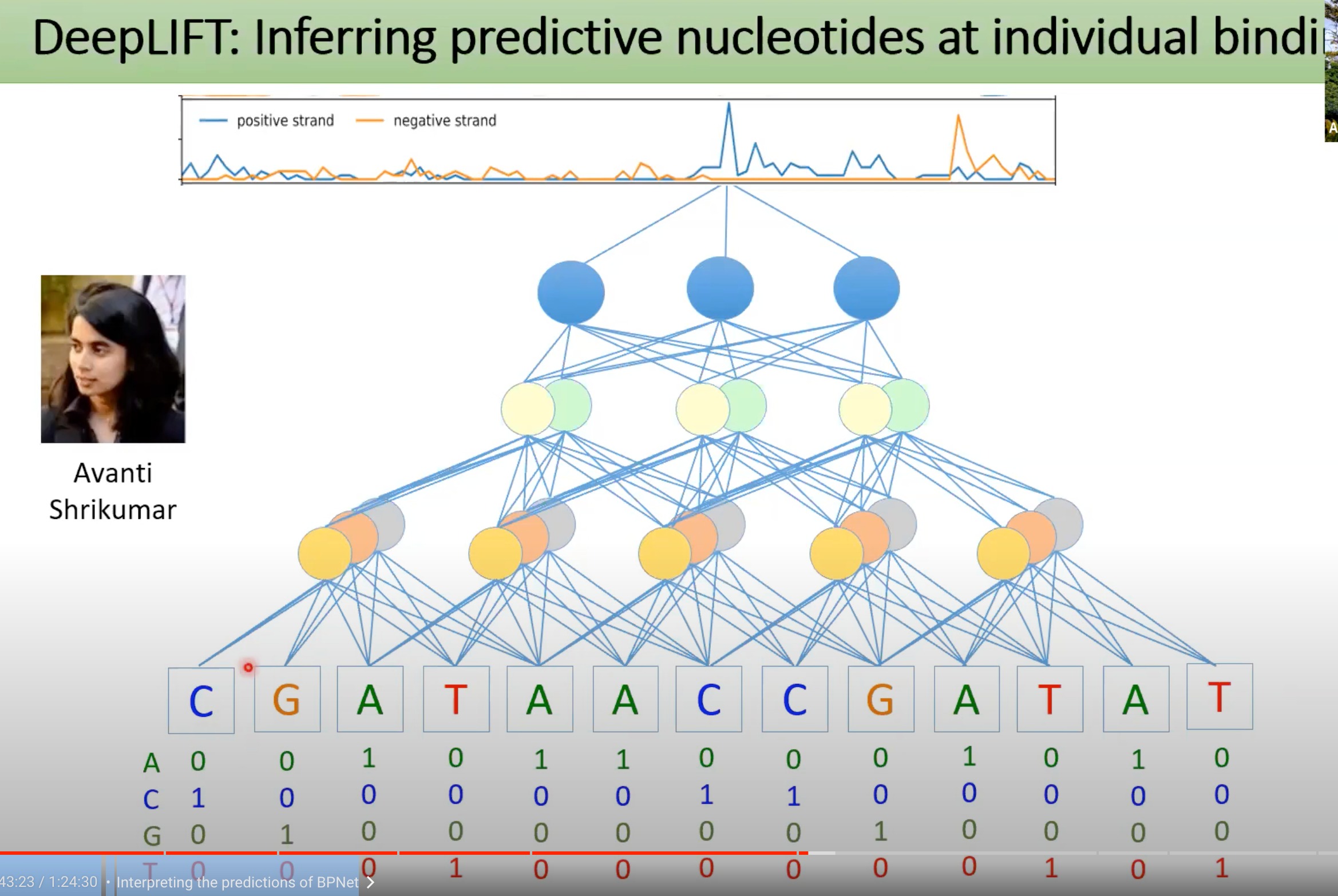
从预测的结果反向去分析每一层神经网络的贡献的大小,进行归因分析,直到输入的每一个核苷酸
如图
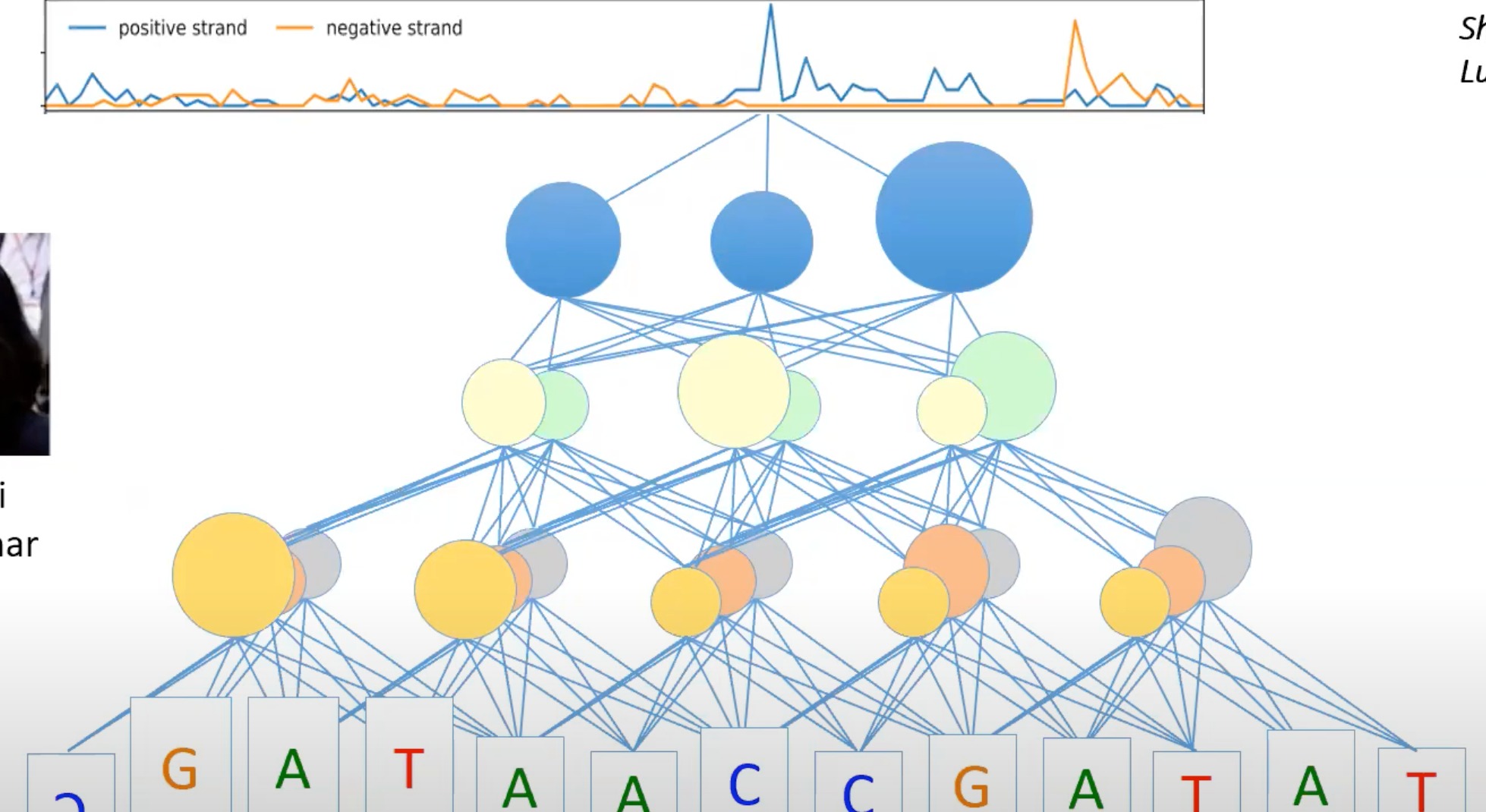
可以分析出每个核苷酸是如何影响的,如何根据context进行预测
- 四种不同转录因子在特定DNA序列区域的结合亲和性
- 可以对基因组中的每个增强子都进行这样的操作
- 这些转录因子喜欢结合到增强子区域
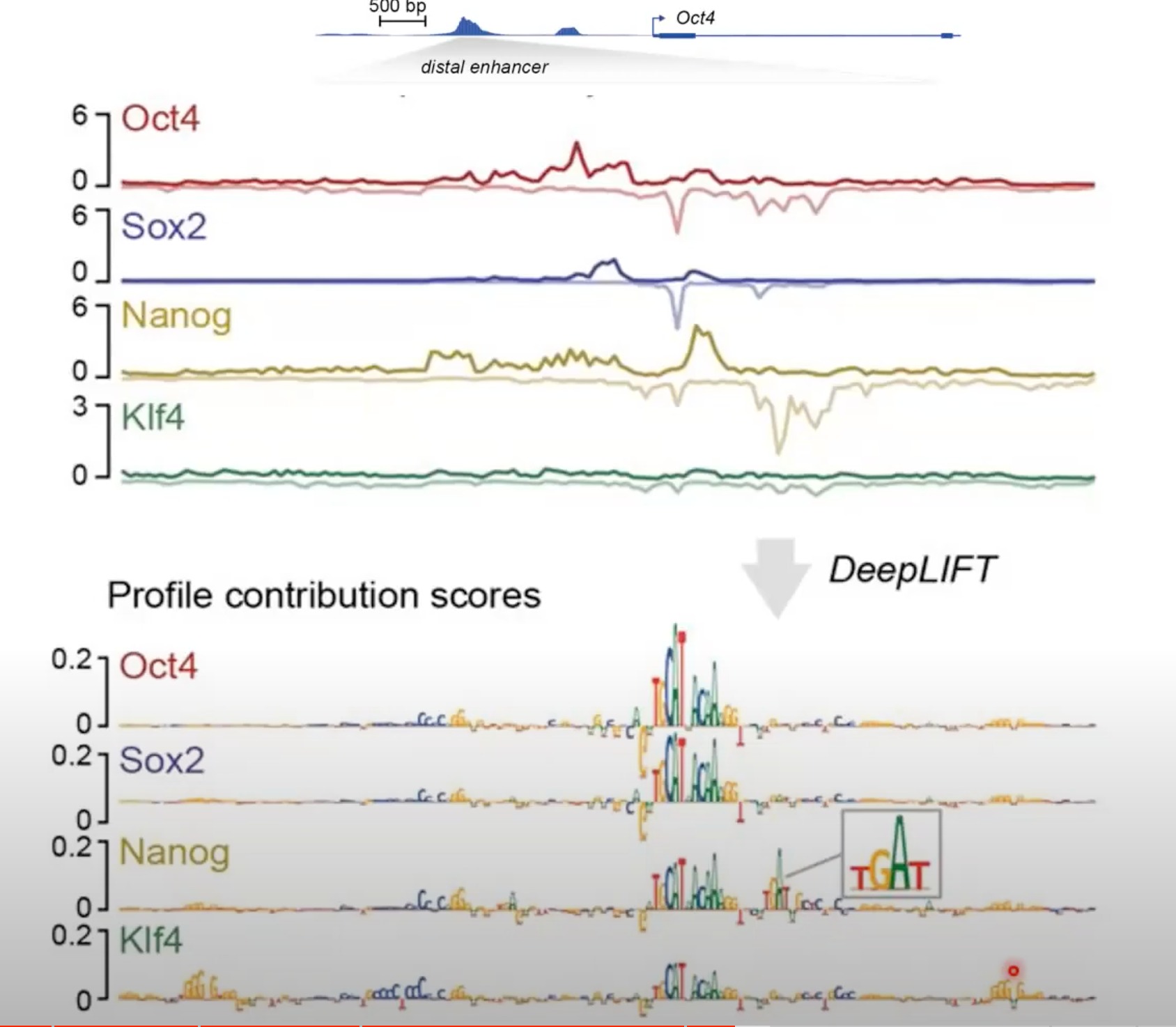
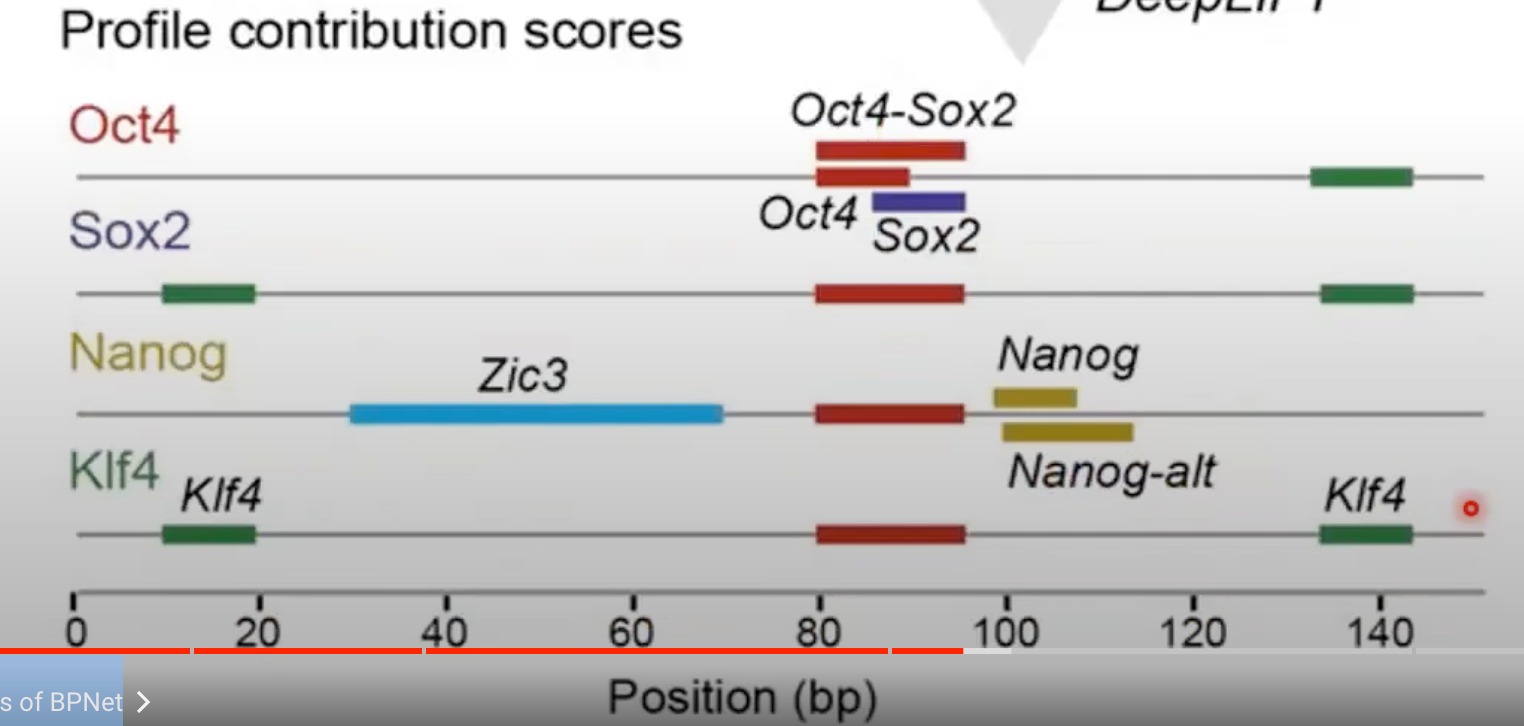
可以看出,对于某个转录因子来说,其有对应的motifs。
而其中的基因调控是十分复杂的,比如:
- 转录因子的协同作用: Oct4和Sox2结合后会形成一个Oct4-Sox2复合体,该复合体有自己独特的DNA结合模式。
- 结合位点的相互作用: 在某些情况下,一个转录因子的结合可能会增加或减少另一个转录因子结合到相邻位点的可能性。这种相互作用可能与DNA的三维结构和染色质的状态有关。
- 染色质的开放性: 当一个转录因子结合到某个区域时,它可能会引起染色质的开放,从而为其他转录因子提供结合的机会。
- 转录因子的多样性
第二张图我还是有一点疑惑的,为什么一个转录因子的线上会有不属于这个因子的小长方形:
我的理解(很有可能错):我们先通过模型的反推得到了哪些motifs是对于这个蛋白来说是贡献大的。我们就将这一小段区域的这个序列跟这个蛋白相关联。然后我们再针对某一个蛋白去看他的chip-seq的结果。我们就可以发现,如果在这个蛋白的这一段区域表达量高。那就说明。他跟这个区域的motifs是相关的
反正意思应该是在某一个转录因子的线上的某个位置有另一个转录因子的结合模式,对当前的转录因子结合有重要贡献,但我不知道怎么测出来的。
以上的这些也许对于研究单个序列的特性还好,但是对于全基因组范围内的还是不够看,所以又开发了一个
2 TF-MoDISCO
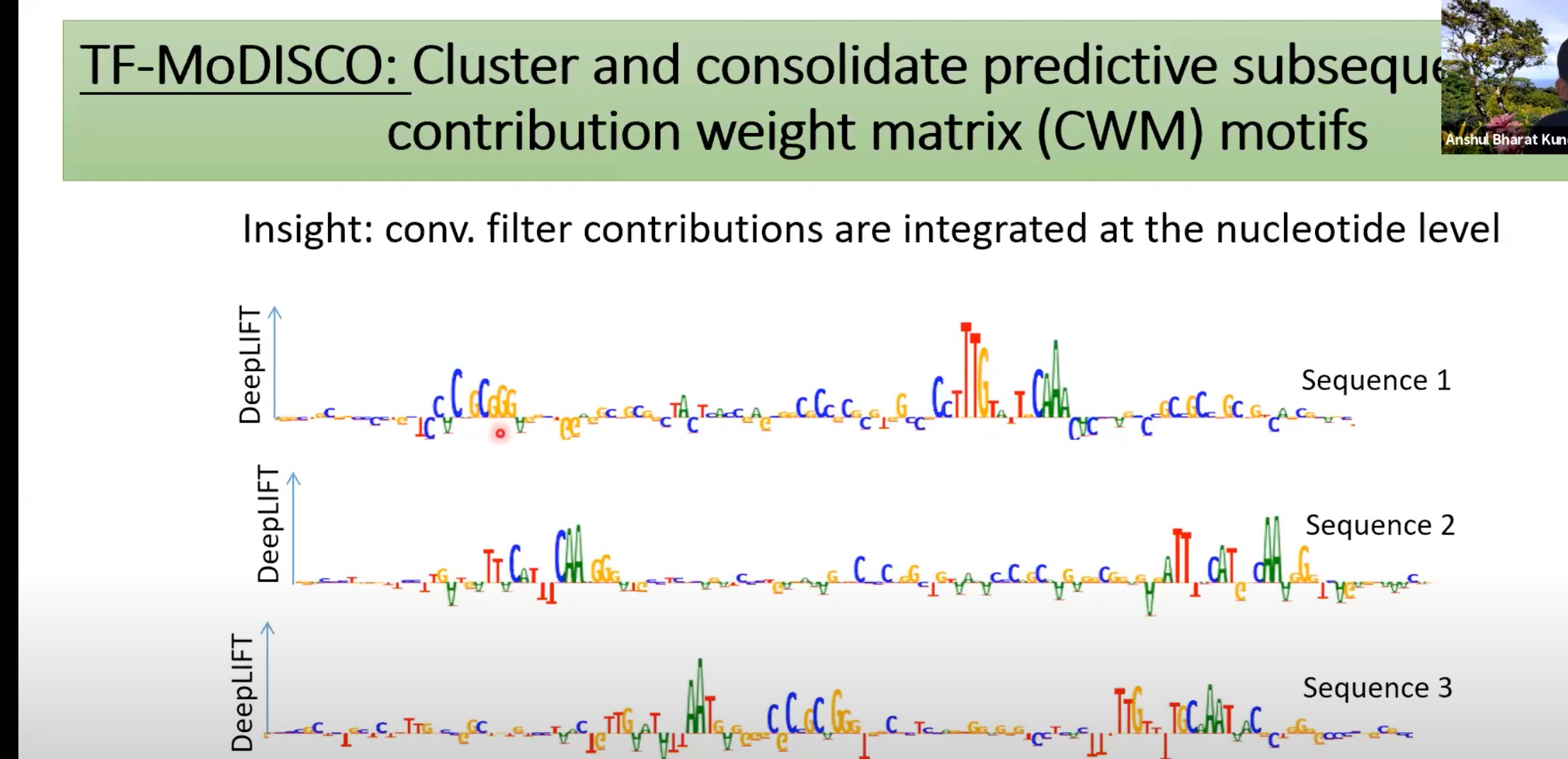
通过DeepLIFT模型,识别和聚类那些对模型预测起到关键作用的motifs
整个多个测得的序列上面的motif

该转录因子最有可能结合的DNA区域
-
CWM & PFM: 这些是模体的不同表示方法。CWM表示模体中每个位置的贡献,而PFM表示每个位置的核苷酸频率。
-
Average contribution score: 表示了每个motif对模型预测的平均贡献。
-
ChIP-nexus footprint: 在实际实验中观察到的转录因子结合模式
-
motifs per region: 这个直方图展示了每个调控区域中模体的数量。
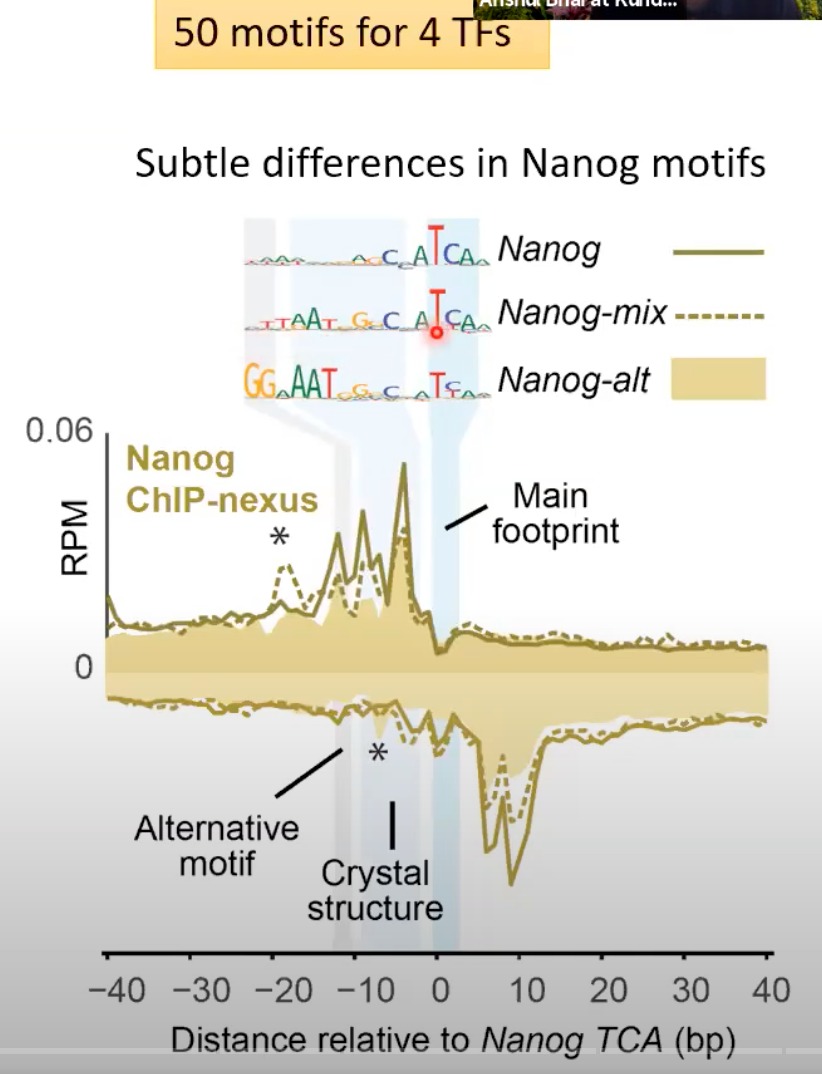
3 motif syntax derived TF cooperativity
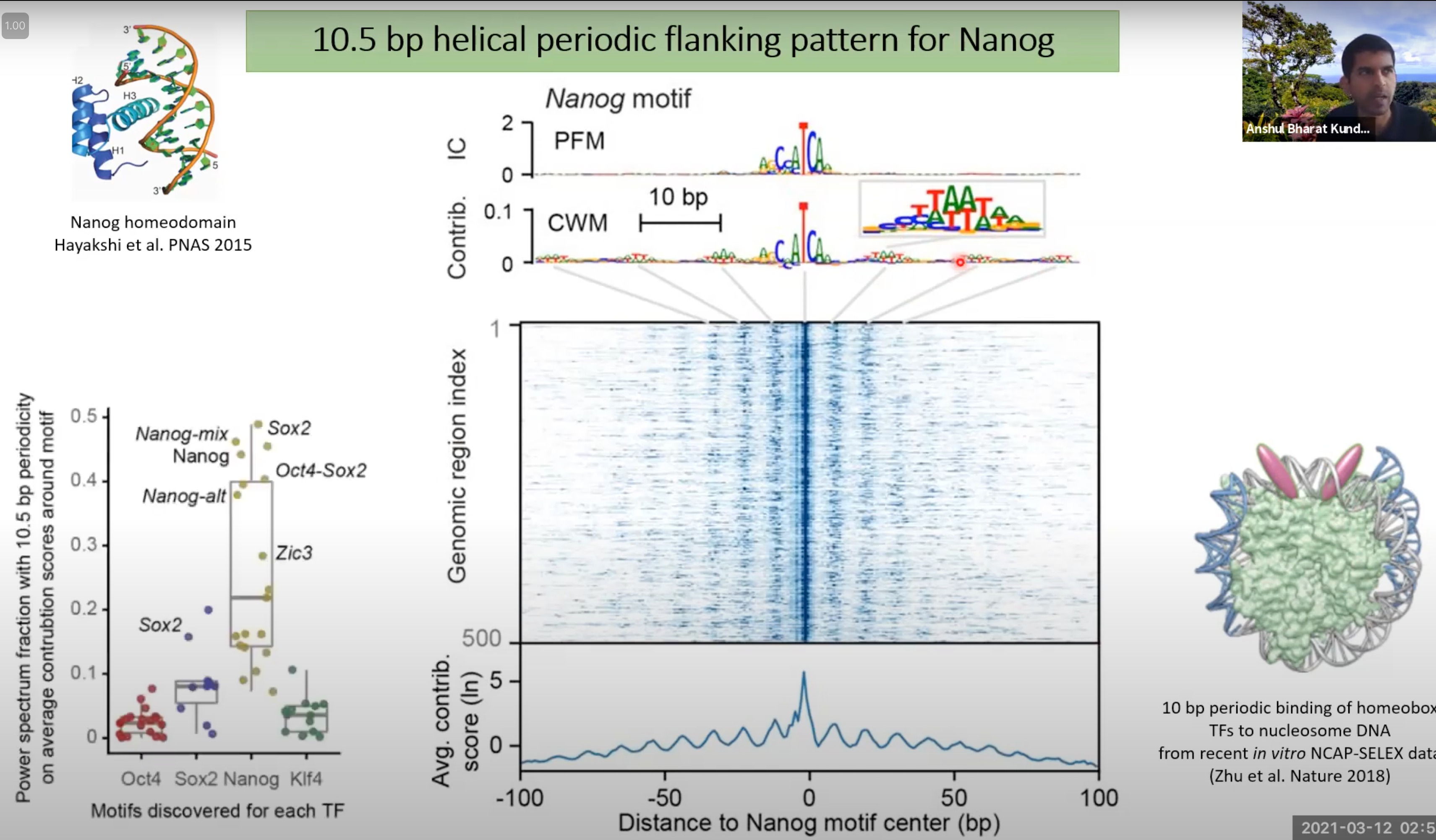
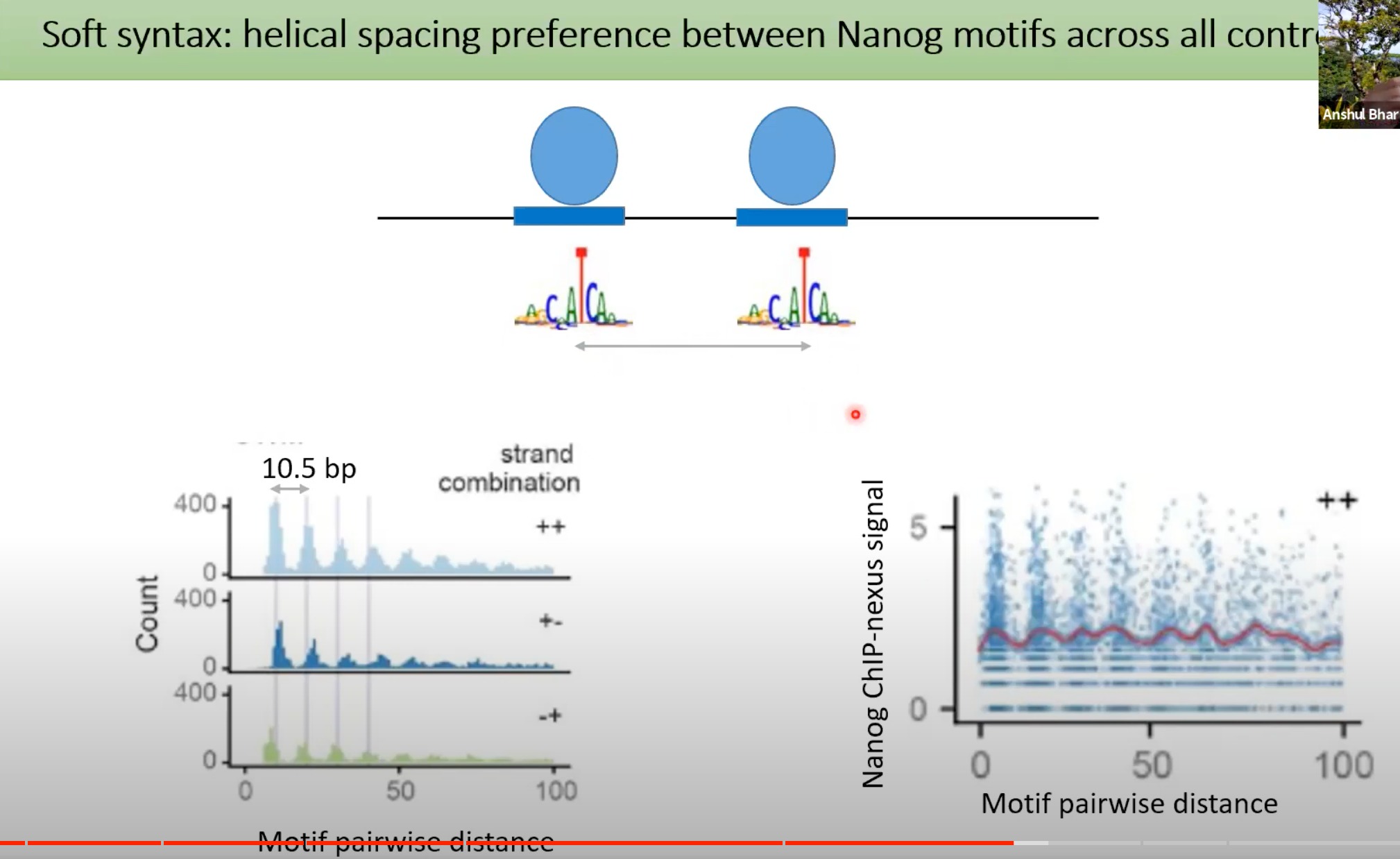
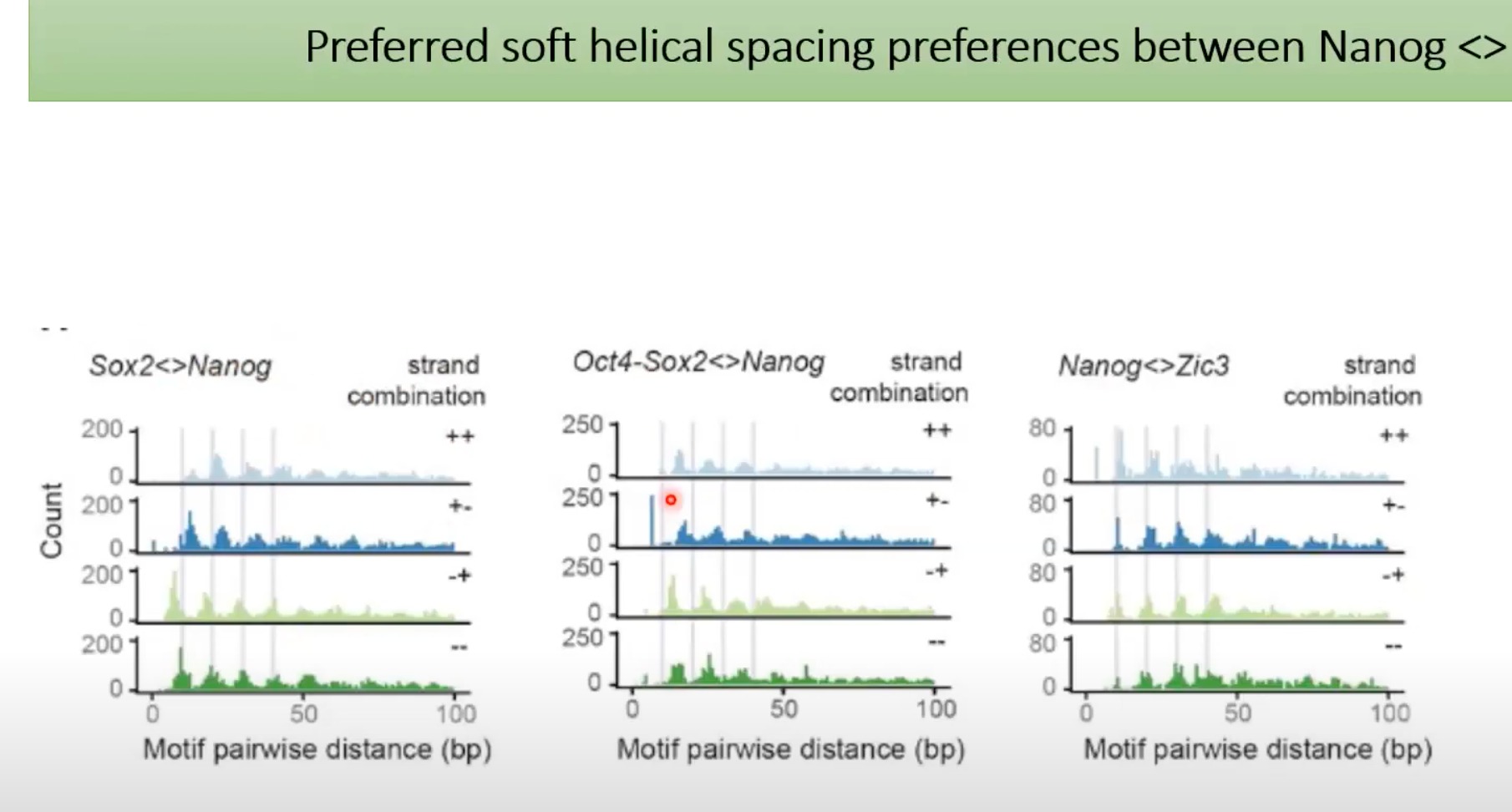
10.5 bp是DNA双螺旋每转大约的碱基对数。所以,当我们看到10.5 bp的周期性,这通常意味着转录因子或其他蛋白质与DNA的某个特定部位结合,并导致这种周期性模式。
-
Nanog的结合模式在DNA上呈现出10.5 bp的螺旋周期性。这意味着与Nanog结合的DNA区域会有一种重复的、周期性的模式。
-
那个二维图,展示了大量基因组区域中Nanog motif的存在情况。深色的线表示Nanog motif在该区域的结合强度较高。
-
可以看出比较深的蓝色线之间,每个的间隔是10.5bp
-
一个特定的motif(如Nanog motif)在多个位置的出现。所以,基本上每个深色的蓝线代表的是相似或相同的motif。但是,因为每个位点的周围环境可能略有不同,所以每个位点上的motif可能会有些许的变化。
-
-
右下角的实验论文证明了10.5bp周期性
-
左下角的图:power spectrum图,用于表示数据中周期性模式的强度。它可以帮助我们检测并量化某种周期性模式在数据中的存在
- 这里的y轴表示周期性的强度,而x轴代表了不同的转录因子。如果一个转录因子的结合模式在DNA上有10.5 bp的周期性,那么该转录因子在图上的点会有较高的y值。简而言之,这个图是用来显示哪些转录因子的结合具有10.5 bp的周期性。
- 通过“motif syntax”来推断不同蛋白质之间的"causal" directional cooperative influence
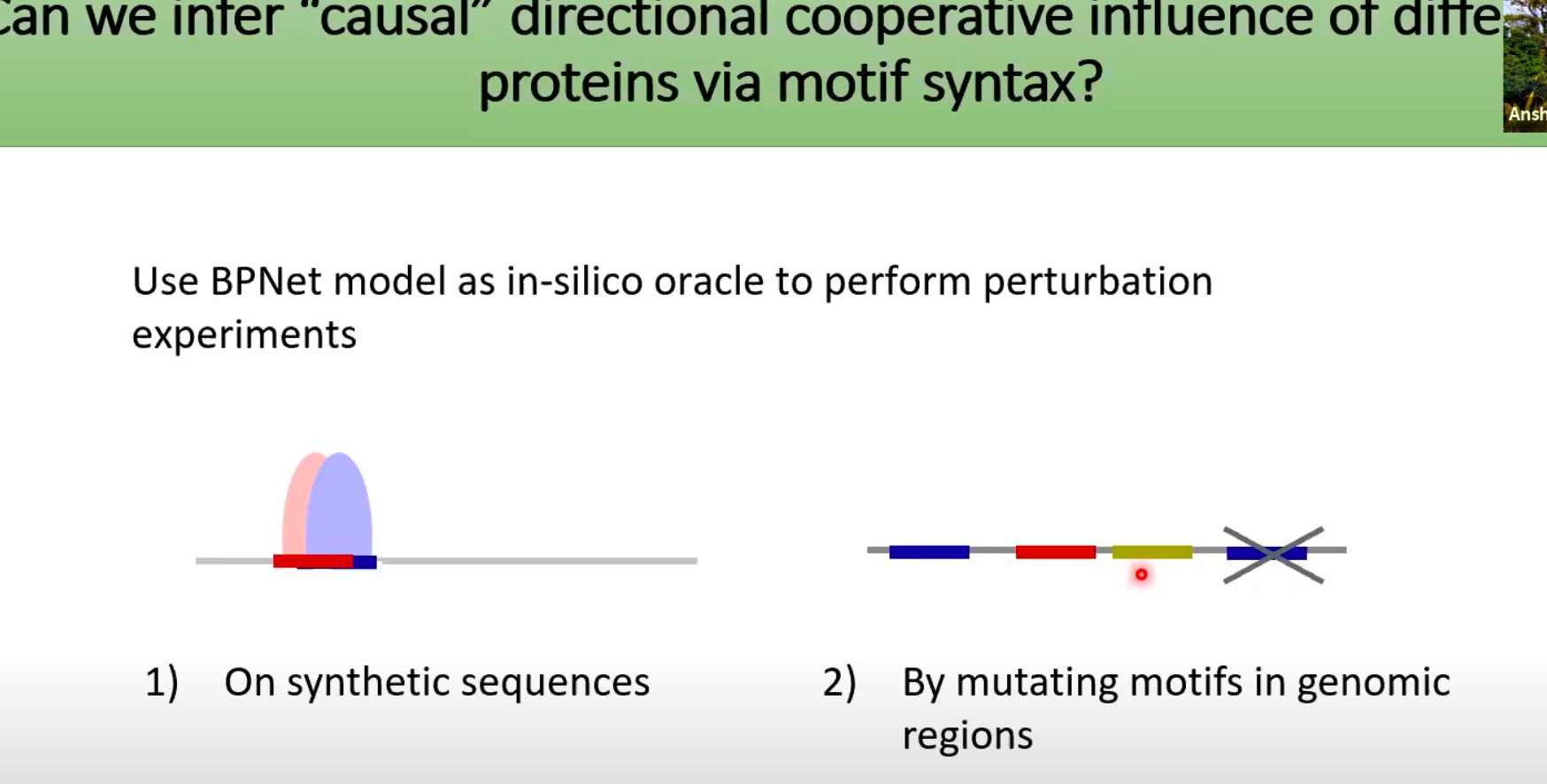
使用BPNet作为一个虚拟的“预言家”来执行扰动实验
两种扰动方法:
- On synthetic sequences:在合成序列上进行。
- 合成序列指的是在实验室中人为制造的DNA或RNA序列,而不是自然界中存在的序列。
- 通过在这些合成序列上运行BPNet模型,并观察蛋白质如何与这些序列相互作用,研究者可以了解这些motifs如何影响蛋白质的行为。
- By mutating motifs in genomic regions:通过在基因组区域内突变 motifs 进行。
- 选择特定的基因组区域,然后人为地突变其中的motifs
- 通过比较突变前后的序列在BPNet模型中的表现,研究者可以推断这些motifs在蛋白质相互作用中的角色。

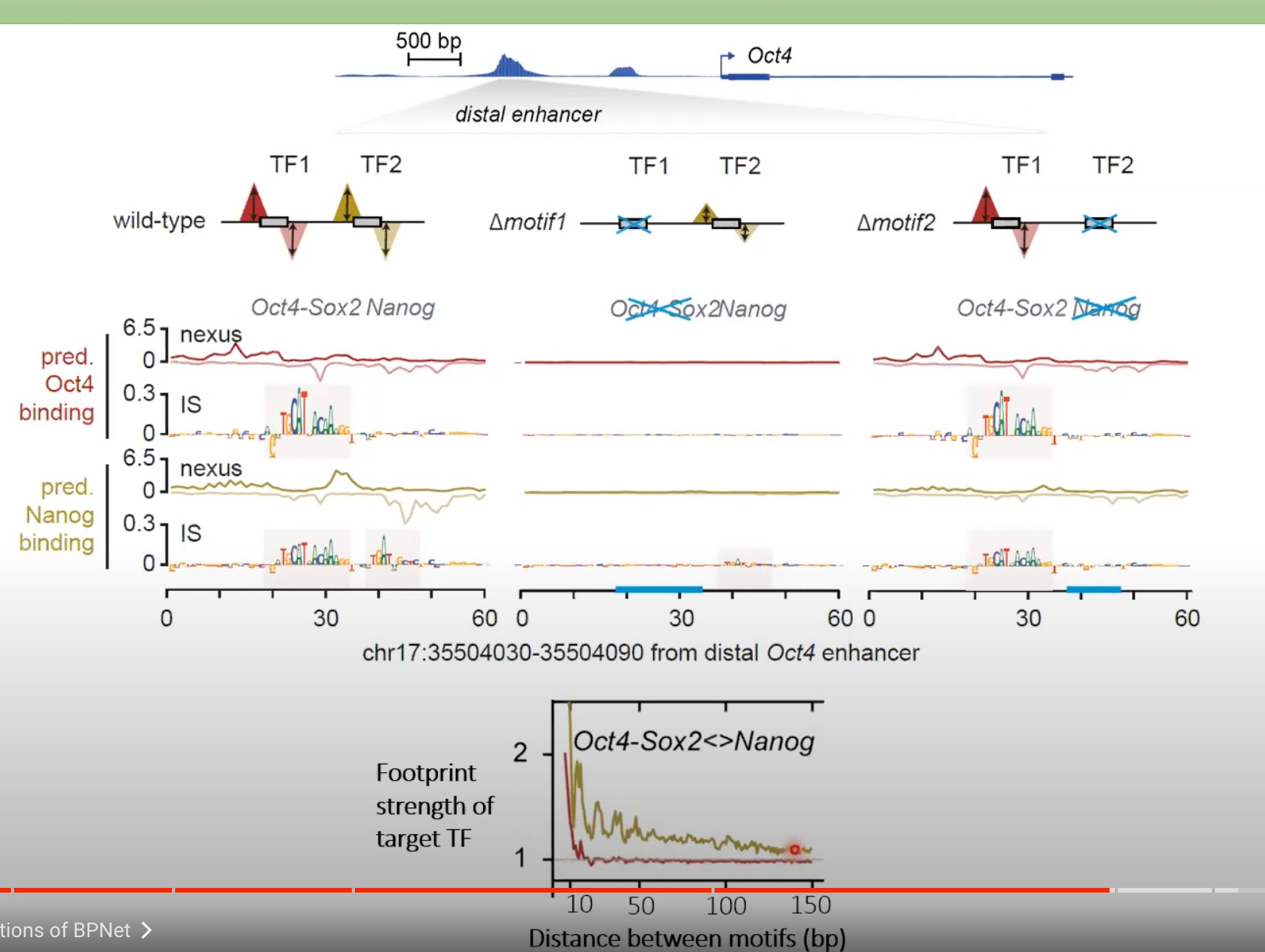
动图:视频52:38处
Oct4和Nanog在合成序列上的合作交互
- 研究者们可以在精确的位置和距离上放置特定的蛋白质结合位点,从而观察蛋白质的结合模式和交互。
- X轴 - 距离 (Distance):X轴表示Oct4-Sox2和Nanog之间结合位点的距离。
- **Y轴 - footprint的改变倍数 **:Y轴显示了蛋白质结合的强度或稳定性的变化。当蛋白质紧密结合到DNA上时,通常会产生一个“footprint”,这是因为结合的蛋白质会阻止其他分子接触该区域。Y轴的值越高,表示结合的稳定性或强度越大。
- Oct4-Sox2 -> Nanog:金色显示了Oct4-Sox2先结合,Nanog后结合时的情况。
- Nanog -> Oct4-Sox2:红线相反
当Oct4-Sox2首先与DNA结合时,它可能更容易促进或稳定Nanog的结合,比之于Nanog先于Oct4-Sox2结合的情况。
- 结构或空间考虑:蛋白质-DNA结合并非只是化学作用,它也受到蛋白质和DNA空间结构的影响。当Oct4-Sox2先结合时,它可能导致DNA的某种空间构象变化,使得Nanog的结合变得更为容易或稳定。
- 可能的生物学意义:在生物学上,这种协同作用可能对某些特定的细胞功能或基因调控过程至关重要。例如,它可能影响基因的启动子活性、基因的表达模式或细胞命运的决策。
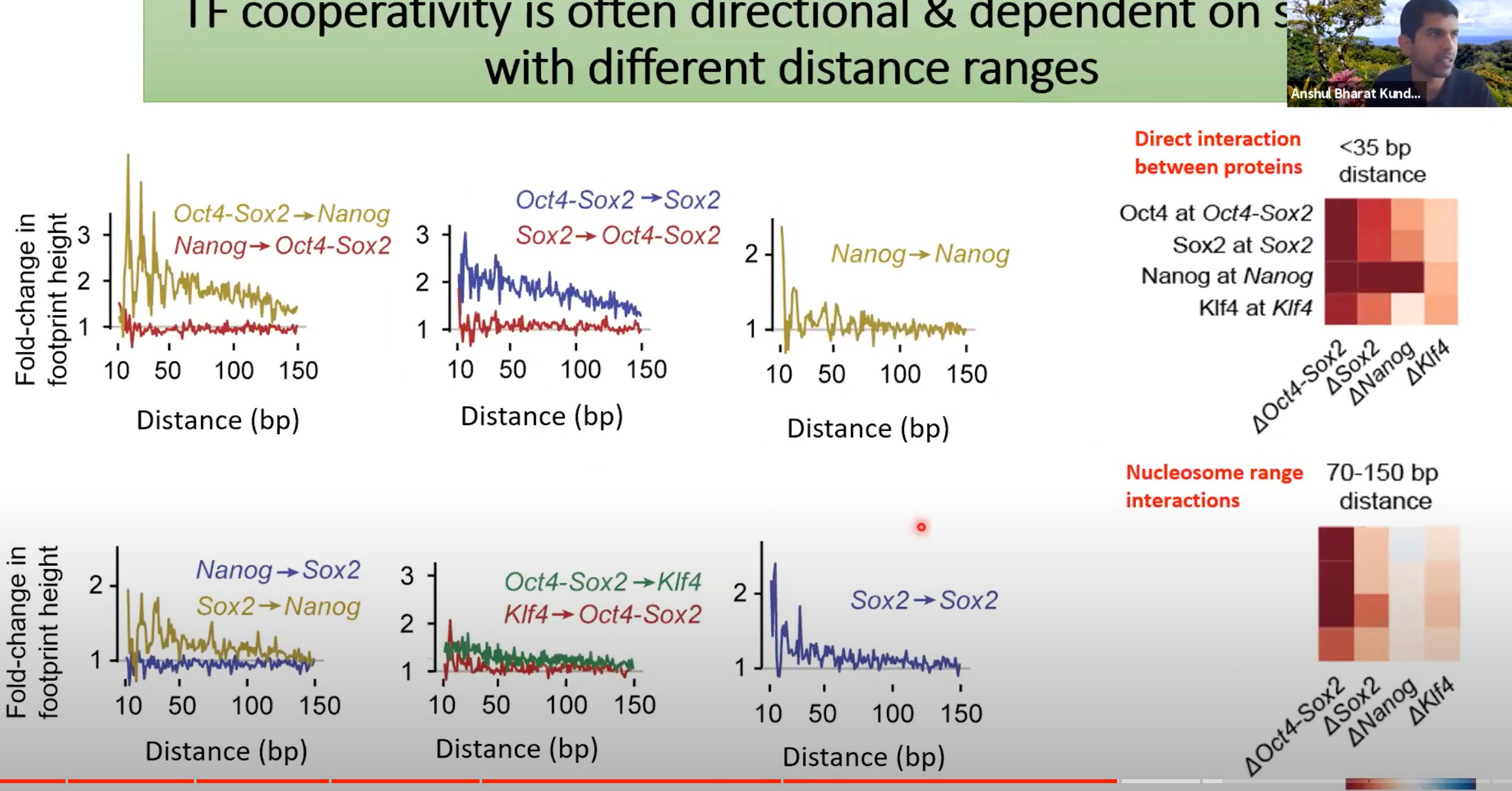
- 不同的转录因子之间的直接互作和核小体范围的互作
- 热力图表示互作强度,Oct4与Oct4-Sox2之间的互作在小于35bp的距离内是非常强的
Experimental validation of BPNet predictions

实验验证:通过CRISPR技术对基因组序列进行的突变如何影响Nanog和Sox2的结合
- “Wt Sox2 motif”和“Mutant Sox2 motif”分别表示Sox2原始的结合模体和突变后的结合模体。
- 当序列发生突变时,蛋白质的结合模式(即观察到的曲线)也发生了改变
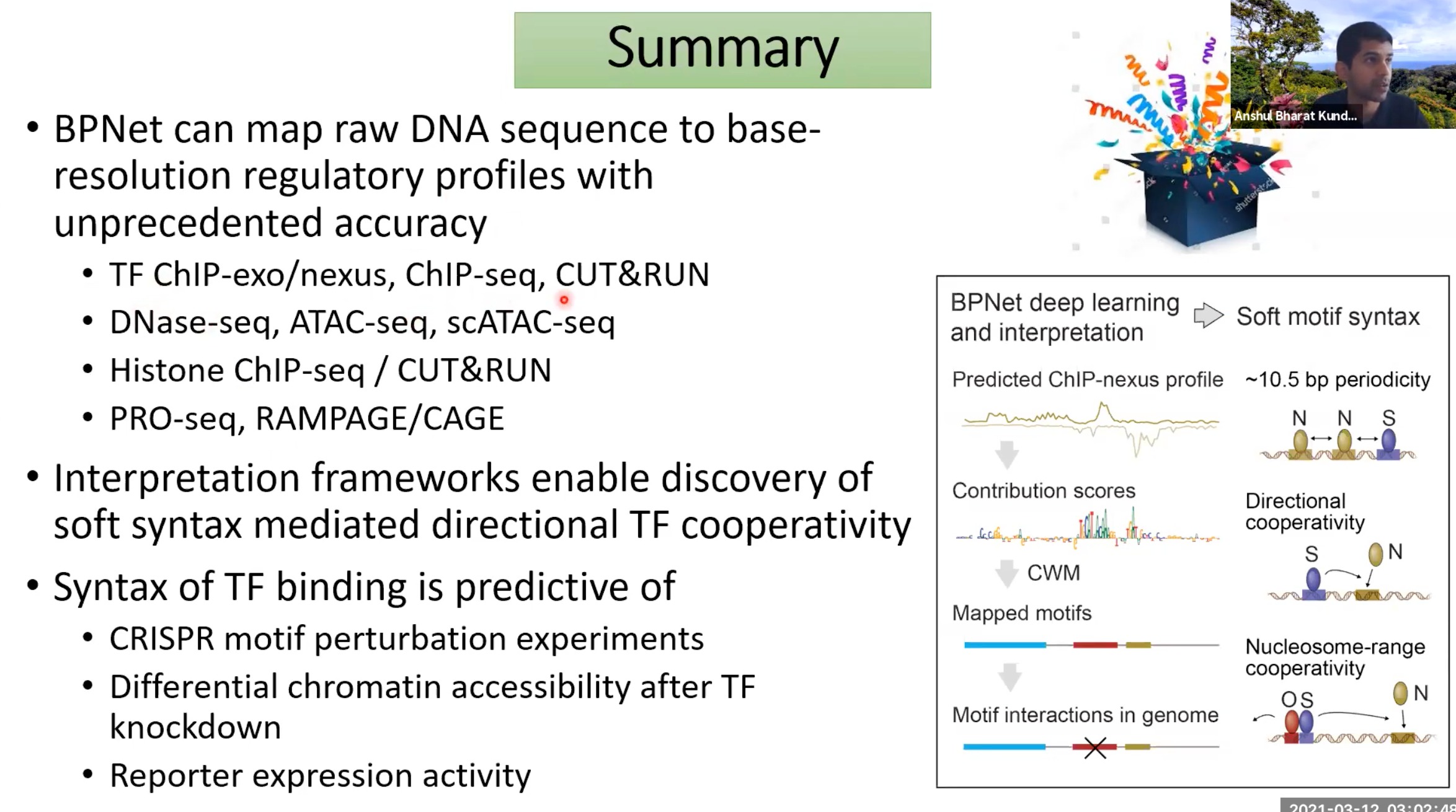
summary